Fragmenstein
Description
NB. The class
Monsterwas formerly calledFragmenstein
The Fragmenstein class places the followup placing algorithm.
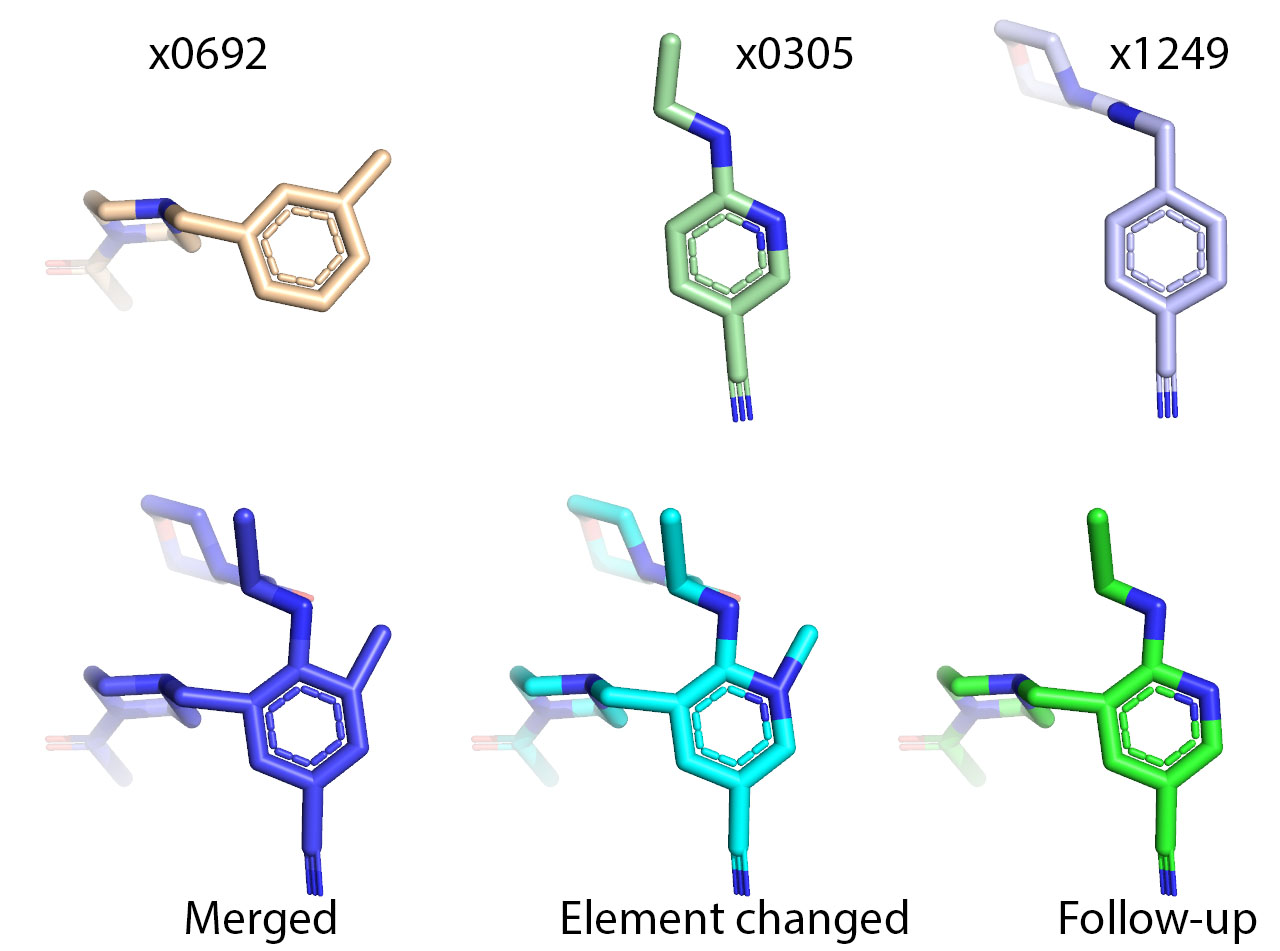
One problem in doing so is mapping atoms from the hits to the followup. Three modes were tested.
The default (recommended) is with not prior merging of the hits.
The three modes rely heavily on mapping one-to-one atomic coordinates of overlapping atoms between hits within a 2Å radius (see Position Mapping code).
The three have different strengths. And which is better depends actually on the dataset. But most likely the no merging mode is like best for most datasets.
The argument merging_mode (full``|``partial``|``none``|``none_permissive``|``off) chooses which is used.
All three of these modes place the atoms of the followup and “project” the missing atoms.
Mapping modes
Full merging
The main documentation for this mode can be found here.
The inspiration hits are merged creating a new merged template and the followup is mapped onto this. This makes it much faster than the unmerged mode, so sliding scale of MCS mappings is done —a very strict MCS mapping is done, then a series of MCS ranging from very lax to more strict are done until one is found that supersets the strictest one.
This was the primary method used, until partial mapping was introduced to overcome the incorrect hit issue.
Pros
Obedient
Can be used to suggest new followups
Pro/Con
Code to stop mapping of unconnected hits (
fuse) disabled/buggy, because of erroneous hits are a bit issue.
Cons
Slower than partial mapping (reason unclear)
Sensitive to too many and to incorrect hits (a COVID moonshot dataset problem)
Ring overlaps can result in really odd rings(∗)
∗ See work in progress for the ring collapsing code which aims to fix this.
Partial mapping
The main documentation for this mode can be found here.
As above but inconsistent hits are excluded.
The “inconsistent” hits (dodgies in the code) are those who when seen in trio of hits do not map consistently.
This speeds up the code and avoids the pitfalls of incorrect hits and too many hits.
Pros
Faster than full
Less prone to too many hits/wrong hits
Cons
often behaves differently than intended by ignoring good hits
No merging
This method (self standing code in core.unmerge_mapper.py) tries to maps each hit to the compound. The maps must satisfy some conditions, two mapped atoms cannot occupy the same 2 Å overlapping position and no bond in the map can be over 3 Å long —if the atoms in between are not placed there is no constraint (which is a problem). This mills through all possible MCS pairing of the various inspiration hits, so the time taken increases to the power of the hits.
The (slower) variant none_permissive allows the mapping of atoms of different kinds assuming the more permissive mapping includes the stricter.
Better at mapping
Better at discarding bad inspiration hits
Slower
Vulnerable to distant erroneous hits
Comparison
For a comparison see three modes compared.
Covalent
If the Chem.Mol has a dummy atom (element symbol: * within RDKit and smiles, but R in a mol file and PDB file) and
a molecule with a single atom is passed to attachment argument, then the covalent linker if absent in the hits is anchored
to that atom.
The default dummy atom can be overridden with Fragmenstein.dummy:Chem.Mol and Fragmenstein.dummy_symbol:str.
Knows its past
The Chem.Mol object will have Chem.Atom objects with the RDKit property _Origin.
This is a json stringified list of reference hit name dot atom index.
fragmenstein.origin_from_mol(mol) will extract these for you.
Also there will be a field,``_StDev``, which is the average of the three axes of
the standard deviation of the positions of the contributing atoms. fragmenstein.origin_from_stdev(mol) extracts them.
Projection
Many atoms may be novel and added to the followup.
Placing
The method called by the class place_from_map, placed the followup using those atoms.
To do a contrained embed in RDKit the reference atoms need to have a good geometry. Consequently, this is not possible. Therefore in the case of sidechains that are novel in the followup a optimised conformer is a superposed against the half placed followup using the 3-4 atoms that are the closest neighbours within the half-placed structure and the side chain position copied from there for each bit.
Imperfect projection
The projection approach is not perfect, but it is not constrained so generally gets fixed without issue during minimisation. This problem is quite apparent in the cases where atoms connecting to the sulfur are added:
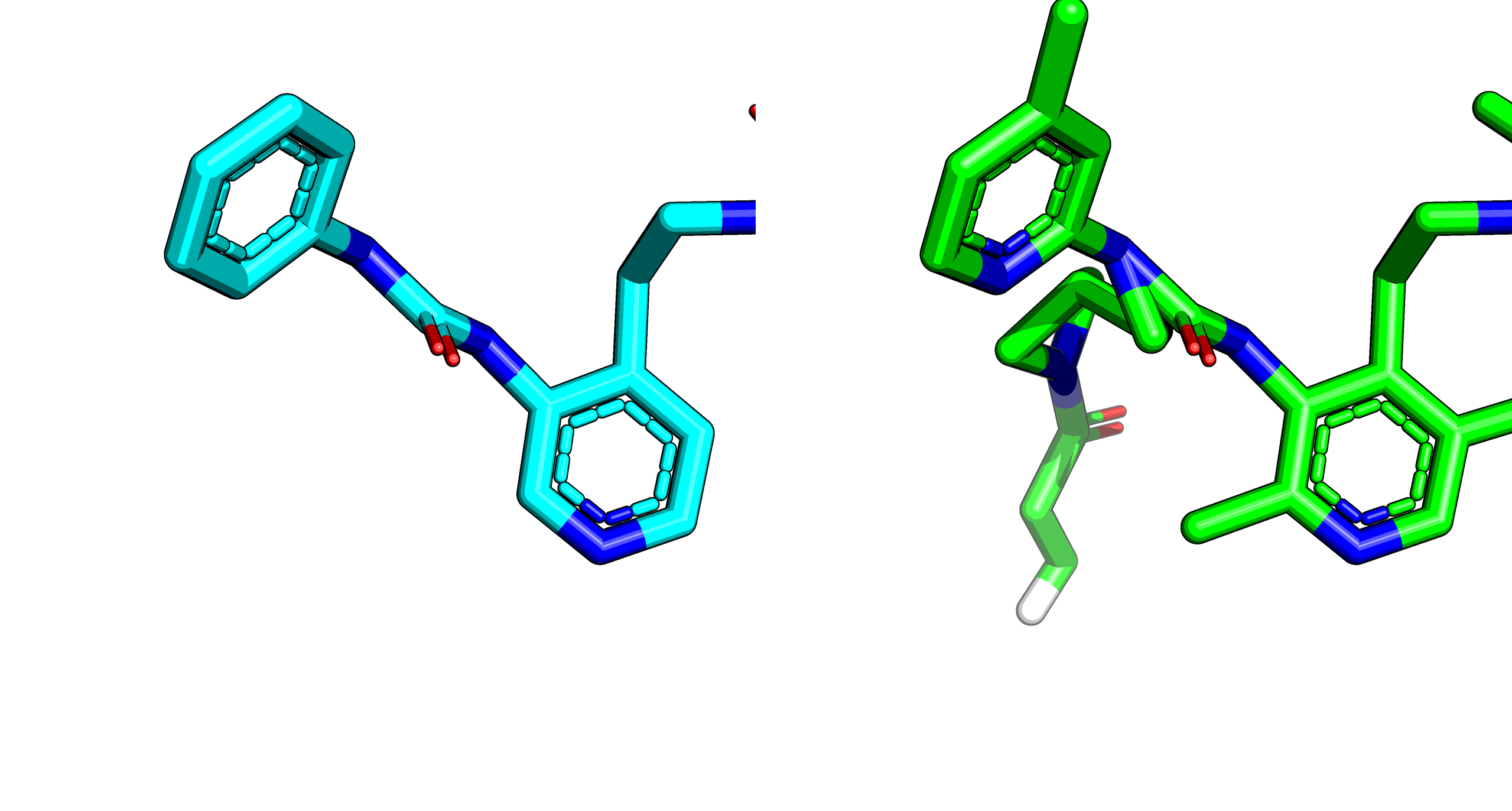
The way the projection is done is via a single conformer.