Generalised potentials
In Rosetta there are three atom types, the primary ones, used by the ref2015 scorefunction and others,
the CHARMM (MM) atom types, used nearly for reference only, and the generalised potential atom types
for the beta_genpot scorefunction.
The first atom types are protein specific, so in the generation of residue types (params files) one may encounter some problematic cases, for example a nitrogen in a tryptophan will be used as a proxy for a nitrile nitrogen. The generalised potentials are aimed at addressing these.
The topology file (params) generation module used by Fragmenstein is rdkit-to-params, which can assign generalised potential atom types.
from rdkit_to_params import Params
# acetonitrile
params = Params.from_smiles('CC#N', generic=True, name='CCN')
print(params.ATOM[2].rtype) # ``NG1`` vs. ``NtrR``
Whereas it is mostly robust, there are some corner cases where it can be problematic. For example, let’s assume we have a covalently bound ligand in a protein with a non-canonical amino acid. These will independently cause issues when read from a PDB file, which is the primary was Fragmenstein operates (primarily to allow inter-operability).
Startup:
import pyrosetta_help as ph
import pyrosetta
extra_options= ph.make_option_string(no_optH=False,
ex1=True,
ex2=True,
gen_potential=True,
score=dict(weights='beta_genpot_cart'),
#mute='all',
ignore_unrecognized_res=False, # raise error if unrecognized
load_PDB_components=False,
ignore_waters=False)
# weights can be one of ['beta_genpot', 'beta_genpot_cart', 'beta_genpot_cst', 'beta_genpot_soft']
pyrosetta.init(extra_options=extra_options)
# no idea why I get this, but it is not an issue:
# core.init.score_function_corrections: [ WARNING ] Flag -beta_nov16 is set but either -weights are also specified or this is being used with the genpot score function. Not changing input weights file!
# core.init.score_function_corrections: [ WARNING ] Flag -gen_potential is set but -weights are also specified. Not changing input weights file!
scorefxn = pyrosetta.get_fa_scorefxn()
assert scorefxn.get_name() == 'beta_genpot_cart'
NB. One can only declare a beta_genpot scorefunction if the option gen_potential is true and at first instantiation
of pyrosetta. This will not work:
pyrosetta.init()
pyrosetta.rosetta.basic.options.set_boolean_option('corrections:gen_potential', True)
pyrosetta.create_score_function('beta_genpot_cart')
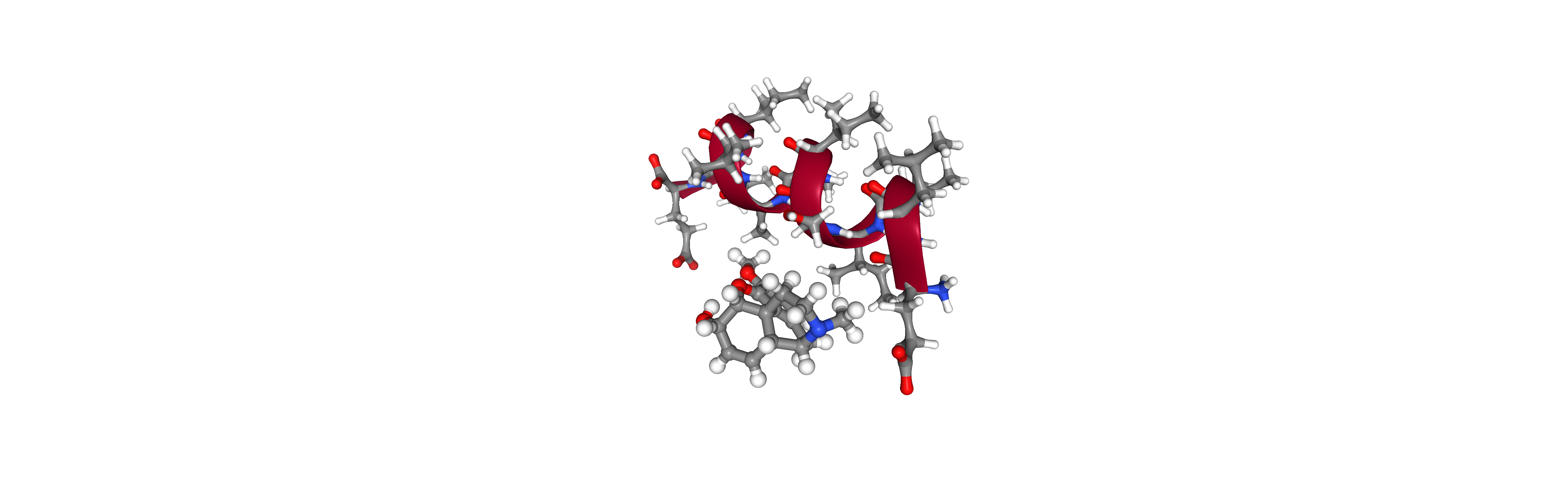
A simple case of a protein and a ligand works fine, either constructed or read from file.
# ## Prep
pose = pyrosetta.pose_from_sequence('ELVISISALIVE')
ph.make_alpha_helical(pose)
codeine_smiles = 'CN1CC[C@]23[C@@H]4[C@H]1CC5=C2C(=C(C=C5)OC)O[C@H]3[C@H](doc_C=C4)O'
params = Params.from_smiles(codeine_smiles, generic=True, name='COD')
codeine_pose = params.to_pose()
pyrosetta.rosetta.core.pose.append_pose_to_pose(pose, codeine_pose, new_chain=True)
# ## minimise
cycles=3
relax = pyrosetta.rosetta.protocols.relax.FastRelax(scorefxn, cycles)
movemap = pyrosetta.MoveMap()
movemap.set_chi(True)
movemap.set_bb(True)
movemap.set_jump(True)
relax.set_movemap(movemap)
relax.minimize_bond_angles(True)
relax.minimize_bond_lengths(True)
relax.apply(pose)
# ## round trip
params.dump('COD.gp.params')
pose.dump_pdb('test.pdb')
qose = ph.pose_from_file('test.pdb', ['COD.gp.params'])
relax.apply(qose)
# ## view
import nglview as nv
view = nv.show_rosetta(pose)
view.add_representation('hyperball')
view.download_image('codeine.png')
view
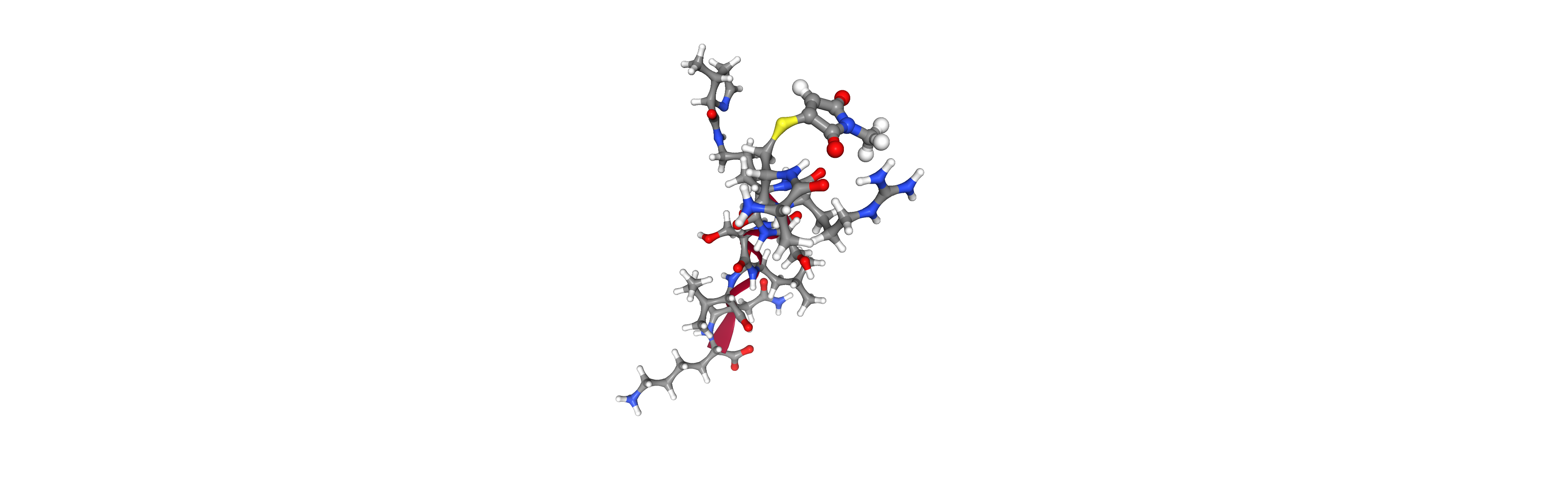
However, things get hairy with more complex cases: non-canonical amino acids and/or covalent ligands read from file.
# ## peptide with NCAA
pyrrolysine_smiles = 'O=C(NCCCC[C@@H](doc_C(=O)*)N*)[C@@H]1/N=C\C[C@H]1C'
params = Params.from_smiles(pyrrolysine_smiles, generic=True, name='PYR')
params.BACKBONE_AA.append('LYS') # as BACKBONE_AA is a singleton it overrides
params.IO_STRING.data[0].name1 = 'O' # equivalent to "appending" a new value
params.dump(f'PYR.gp.params')
pose = params.to_polymeric_pose(sequence='ACROSSLINK') # the alanine is to avoid an N-terminal patch
ph.make_alpha_helical(pose)
# ## covalent ligand
# the product of the nucleophilic addition of thiol to maleimide is a succinimide,
# the amine reactive succinimide is N-hydroxylsuccinimide (NHS)
xmethylsuccinimide_smiles = 'C1=C(*)C(=O)N(C)C1=O'
params = Params.from_smiles(xmethylsuccinimide_smiles, generic=True, name='XMS')
params.rename_atom(' C2 ', ' CX ')
params.dump(f'XMS.gp.params')
xmethylsuccinimide_pose = params.to_pose()
# ## merge and link
pyrosetta.rosetta.core.pose.append_pose_to_pose(pose, xmethylsuccinimide_pose, new_chain=True)
pyrosetta.rosetta.protocols.simple_moves.MutateResidue(target=2, new_res='CYS:S-conjugated').apply(pose)
pose.conformation().declare_chemical_bond(seqpos1=2, atom_name1=' SG ', seqpos2=11, atom_name2=' CX ')
assert 'LINK' in ph.get_pdbstr(pose)
print(f'The ligand {pose.residue(11).type().name()} has {pose.residue(11).connect_map_size()} connections '
f'which are correctly defined: {not pose.residue(11).connect_map(1).incomplete()}\n'
f'The residue {pose.residue(2).type().name()} has {pose.residue(2).connect_map_size()} connections')
This works.
The ligand XMS has 1 connections which are correctly defined: True
The residue CYS:S-conjugated has 3 connections
The cysteine is CYS:S-conjugated as opposed to SidechainConjugation because it just does not work otherwise
even in standard potentials mode.
However, on minimisation things go south:
relax.apply(pose)
[ ERROR ] Issue getting stub for atom atomno= 26 rsd= 4 -- possibly due to degenerate/colinear atoms
which results in the RunTimeError
Cannot create normalized xyzVector from vector of length() zero. Error occurred when trying to normalize the vector between points A and B. A=[1.506000,-5.628818,-21.391746], B=[1.506000,-5.628818,-21.391746].
Residue 4 is the NCAA. So let’s make it a lysine:
pyrosetta.rosetta.protocols.simple_moves.MutateResidue(target=4, new_res='LYS').apply(pose)
relax.apply(pose)
Nope:
ERROR: Error in core::scoring::methods::RamaPreProEnergy::residue_pair_energy(): The RamaPrePro term is incompatible with cyclic dipeptides (as is most of the rest of Rosetta).
Note that if the covalent bond is not there, it minimises fine most of the time with pyrrolysine. Also, reading from file does not work:
pose = ph.pose_from_file('test2.pdb', ['PYR.gp.params', 'XMS.gp.params'])
[ WARNING ] Patch SidechainConjugation implies it can apply to residue type CYZ, but actually applying it fails.
This non–S-conjugated pose will give the error:
ERROR: No structure mods allowed during scoring!
When any of these operations are done with ref2015 scorefunction (and parameterised with generic=False) it works fine.